How GIVLAARI® (givosiran) Works
What is GIVLAARI?
GIVLAARI is a double-stranded, small interfering RNA (siRNA) therapeutic specifically targeting ALAS1 mRNA, reducing ALAS1 mRNA levels and leading to reductions in urinary ALA and PBG.1,2
Watch how GIVLAARI works
See how GIVLAARI targets ALAS1 mRNA for degradation, leading to a reduction in levels of ALA and PBG.1,2
See Long-term GIVLAARI Efficacy Data
GIVLAARI is indicated for the treatment of adults with AHP. GIVLAARI can be considered for treating a patient regardless of their attack history
Patients with acute hepatic porphyria (AHP) have an enzyme deficiency in the heme biosynthesis pathway3
ALAS1 upregulation leads to the accumulation of neurotoxins that cause damage across the body4
Disease triggers, such as infections or certain medications, can induce upregulation of ALAS1 in the liver3
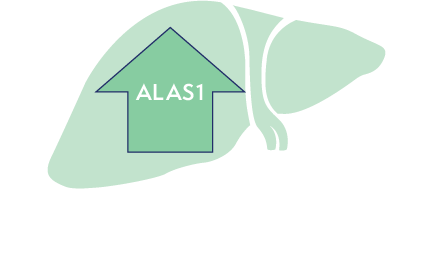
Upregulation of ALAS1 leads to the accumulation of neurotoxic intermediates ALA and PBG in the liver4,5
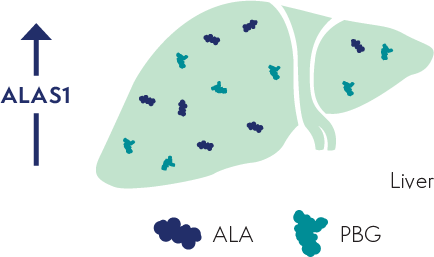
ALA and PBG are
released into circulation, thereby
causing neurotoxic effects4,5
ALA
PBG
ALA=delta-aminolevulinic acid; ALAS1=delta-aminolevulinic acid synthase 1; mRNA=messenger RNA; PBG=porphobilinogen.
GIVLAARI specifically targets ALAS1, key to the pathophysiology of AHP1
GIVLAARI targets ALAS1 mRNA for degradation in the liver1,3
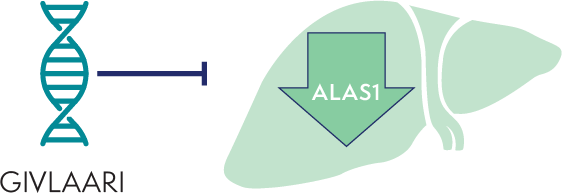
ALAS1 mRNA degradation reduces the production of the neurotoxic intermediates ALA and PBG1,5
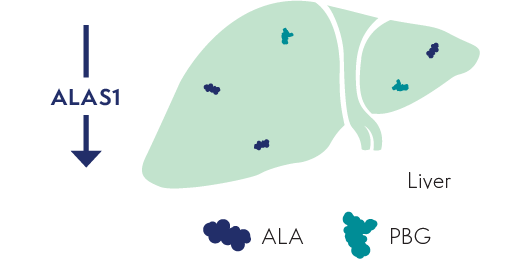
Less ALA and PBG are released into circulation. Reductions of ALA and PBG have been associated with fewer AHP attacks1,2,5
ALA
PBG
ALA=delta-aminolevulinic acid; ALAS1=delta-aminolevulinic acid synthase 1; mRNA=messenger RNA; PBG=porphobilinogen.
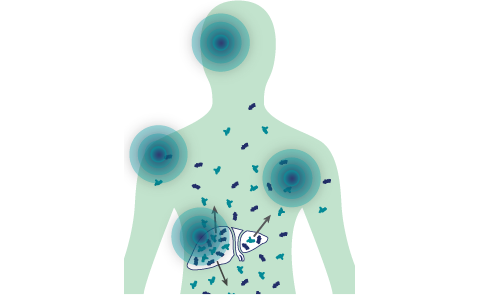
Neurotoxic effects of ALA and PBG are factors associated
with AHP attacks and other disease manifestations6,7
Long-term GIVLAARI treatment reduced ALA and PBG by over 90%8
Urinary ALA levels8

Urinary PBG levels8

*The determination of the ULN for ALA (1.5 mmol/mol of creatinine) was based on samples obtained from 150 healthy persons.
†Data for GIVLAARI 1.25 mg/kg and 2.5 mg/kg in the OLE period were pooled.
‡The determination of the ULN for PBG (0.14 mmol/mol of creatinine) was based on samples obtained from 150 healthy persons.
ALA=delta-aminolevulinic acid; DB=double blind; OLE=open-label extension; PBG=porphobilinogen.
Reductions through the ENVISION 6-month double-blind period1,9
- Reductions in urinary ALA and PBG were seen with GIVLAARI 2.5 mg/kg at Day 14, the first time point measured
- 14 days after the first dose of GIVLAARI, median reductions from baseline in urinary ALA and PBG were 84% and 75%, respectively
- Maximal reductions in ALA and PBG levels were achieved around Month 3 with GIVLAARI 2.5 mg/kg, with median reductions from baseline of 94% for ALA and 95% for PBG, and were sustained thereafter with repeated once-monthly dosing
Reductions in the ENVISION open-label extension (OLE) period10
- In patients who continued treatment with GIVLAARI in the OLE period, reductions in urinary ALA and PBG were sustained through Month 36
- 92.6% median reduction (Q1, Q3: 96.0%, 88.3%) and 95.9% median reduction (Q1, Q3: 99.2%, 90.7%) from baseline in urinary ALA and PBG, respectively
- In patients who crossed over from placebo to GIVLAARI in the OLE period (a total of 30 months of treatment with GIVLAARI):
- 92.0% median reduction (Q1, Q3: 94.9%, 86.9%) and 94.2% median reduction (Q1, Q3: 98.1%, 85.1%) from baseline in urinary ALA and PBG, respectively, were observed at Month 36
Elevated ALA and PBG are associated with AHP attacks6,7
References: 1. GIVLAARI [prescribing information]. Cambridge, MA: Alnylam Pharmaceuticals, Inc. 2. Sardh E, Harper P, Balwani M, et al. N Engl J Med. 2019;380(6):549-558. 3. Balwani M, Wang B, Anderson KE, et al; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Hepatology. 2017;66:1314-1322. 4. Puy H, Gouya L, Deybach JC. Lancet. 2010;375:924-937. 5. Pischik E, Kauppinen R. Appl Clin Genet. 2015;8:201-214. 6. Szlendak U, Bykowska K, Lipniacka A. Adv Clin Exp Med. 2016;25(2):361-368. 7. Kuo H-C, Huang C-C, Chu C-C, et al. Eur Neurol. 2011;66(5):247-252. 8. Kuter DJ, Bonkovsky HL, Monroy S, et al. Presented at: American Society of Hematology (ASH) 2021 Annual Meeting; December 11-14, 2021. 9. Ventura P, Bonkovsky HL, Gouya L, et al. Liver Int. 2021;00:1-12. 10. Data on file, Alnylam Pharmaceuticals, Inc.