including acute intermittent porphyria (AIP),
SIGNIFICANTLY
REDUCED ATTACKS1,2
An attack can be a turning point—and a consideration to start treatment for patients with AHP3-5
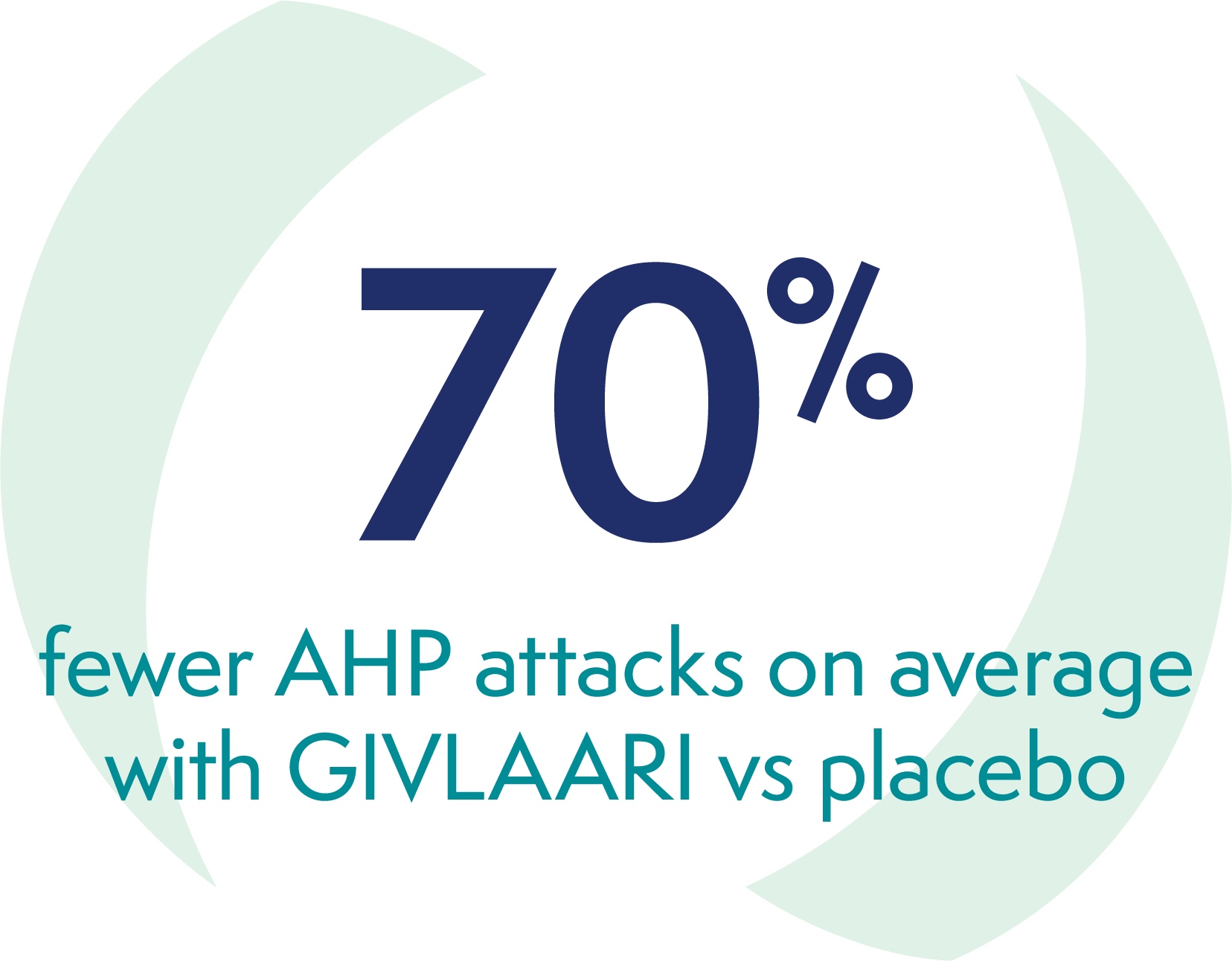
In the 6-month double-blind period of the ENVISION study, patients with AHP receiving GIVLAARI experienced1:
In the 36-month analysis of the ENVISION double-blind and open-label extension (OLE) periods2:

Attack rate ratio: GIVLAARI vs placebo 0.3 (95% CI: 0.2, 0.4; P<0.0001)1
In the OLE period, patients who crossed over from placebo to GIVLAARI had reductions in attacks similar to GIVLAARI patients in the double-blind period.2
- Endpoints in the ENVISION OLE period are exploratory2
In the 6-month double-blind period of the ENVISION study, patients with AHP receiving GIVLAARI experienced1:
Attack rate ratio: GIVLAARI vs placebo 0.3 (95% CI: 0.2, 0.4; P<0.0001)1
In the 36-month analysis of the ENVISION double-blind and open-label extension (OLE) periods2:
In the OLE period, patients who crossed over from placebo to GIVLAARI had reductions in attacks similar to GIVLAARI patients in the double-blind period.2
- Endpoints in the ENVISION OLE period are exploratory2
Attacks were defined as those requiring hospitalization, urgent healthcare visit, or intravenous (IV) hemin administration at home.1
GIVLAARI targets and causes degradation of ALAS1 mRNA, reducing the production of the neurotoxic intermediates ALA and PBG1
ALA and PBG are factors associated with attacks and other disease manifestations of AHP
ALA=delta-aminolevulinic acid; ALAS1=delta-aminolevulinic acid synthase 1; CI=confidence interval; mRNA=messenger RNA; PBG=porphobilinogen.
References: 1. GIVLAARI [prescribing information]. Cambridge, MA: Alnylam Pharmaceuticals, Inc. 2. Kuter DJ, Bonkovsky HL, Monroy S, et al. J Hepatol. 2023;79(5):1150-1158. 3. Kubisch I, Wohmann N, Wissniowski TT, et al. J Clin Med. 2024;13(22):6779. 4. Anderson KE, Lobo R, Salazar D, et al. Am J Med Sci. 2021;362(2):113-121. 5. Simon A, Pompilus F, Querbes W, et al. Patient. 2018;11(5):527-537.
Exceptional insurance access
![[99%] of patients have confirmed access icon](/sites/default/files/inline-images/1.0_Home_PrcntAccess_2.png)
*Coverage may vary for individual and plan. Data as of January 2025.