Efficacy & Safety
ENVISION: The largest interventional study in acute hepatic porphyria (AHP)1,2
ENVISION was a randomized, double-blind, placebo-controlled, multinational phase 3 study in patients with AHP (N=94), with a 30-month open-label extension (OLE) period (N=93)1-3

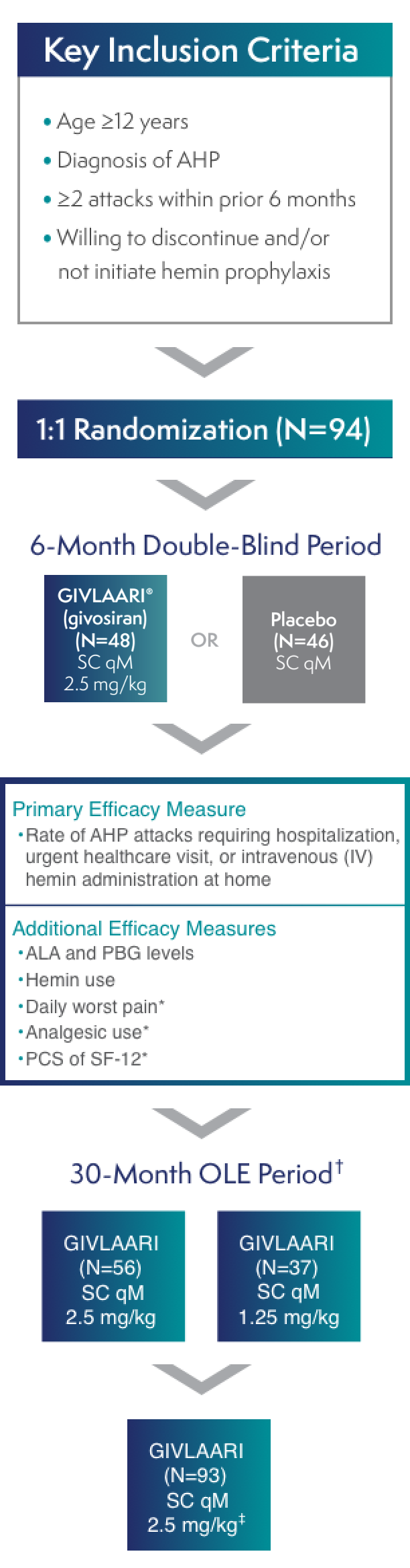
*These measures were in AIP patients only.3
†All endpoints were considered exploratory in the ENVISION OLE period.4
‡A protocol amendment increased the dose for all patients to 2.5 mg/kg once monthly.2
AIP=acute intermittent porphyria; ALA=delta-aminolevulinic acid; PBG=porphobilinogen; PCS=Physical Component Summary; qM=once monthly; SC=subcutaneous; SF-12=12-Item Short-Form Health Survey.
- Patients who experienced an AHP attack during the study were treated according to the local standard of care, which could include IV hemin3
- GIVLAARI was studied in patients experiencing ≥2 attacks in the 6 months prior to study entry to ensure baseline attack rates allowed for a measurable difference in treatment effect on the primary attack rate endpoint5
Patients are not required to have a certain number of attacks to be prescribed GIVLAARI1
| GIVLAARI (N=48) | Placebo (N=46) | Overall (N=94) | |
| Mean age, years (range) | 42 (19, 65) | 36 (20, 60) | 37.5 (19, 65) |
| Female, n (%) | 43 (90) | 41 (89) | 84 (89) |
| Caucasian, n (%) | 39 (81) | 34 (74) | 73 (78) |
| AHP type, n (%) | |||
| – AIP | 46 (96) | 43 (93) | 89 (95) |
| – HCP | 1 (2) | 0 (0) | 1 (1) |
| – VP | 1 (2) | 1 (2) | 2 (2) |
| – No identified mutation* | 0 (0) | 2 (4) | 2 (2) |
| Historical annualized attack rate, median (IQR)† | 8 (4-18) | 7 (4-14) | 8 (4-16) |
| Prior hemin prophylaxis, n (%) | 20 (42) | 18 (39) | 38 (40) |
| Prior chronic opioid use, n (%)‡ | 14 (29) | 13 (28) | 27 (29) |
| Prior chronic symptoms, n (%)§ | 23 (48) | 26 (57) | 49 (52) |
*The 2 patients with acute hepatic porphyria without an identified mutation were considered by the trial investigator to have acute intermittent porphyria on the basis of biochemical analysis.3
†IQR=interquartile range.
‡Use of opioids was defined as chronic if patients reported taking them for porphyria daily, or on most days, when not having an attack.
§Symptoms were chronic if patients experienced symptoms of porphyria daily, or on most days, when not having an attack.
The historical annualized attack rate was calculated as the number of attacks resulting in a composite of hospitalization, a visit to a healthcare facility, or hemin use at home during the 6 months before randomization.3
All eligible patients (93 of 94) enrolled in the OLE period2
Efficacy in the 36-month analysis of the ENVISION double-blind and open-label extension (OLE) periods
In patients with AHP in the ENVISION 6-month double-blind (DB) period,
GIVLAARI® (givosiran) led to a significant reduction in porphyria attacks1
Average rate of porphyria attacks1


Attacks were defined as those requiring hospitalization, urgent healthcare visit, or intravenous (IV) hemin administration at home.1
In a 36-month analysis of patients with AHP,
Long-term GIVLAARI treatment demonstrated sustained attack reduction6
Average number of attacks per 3-month interval5,6
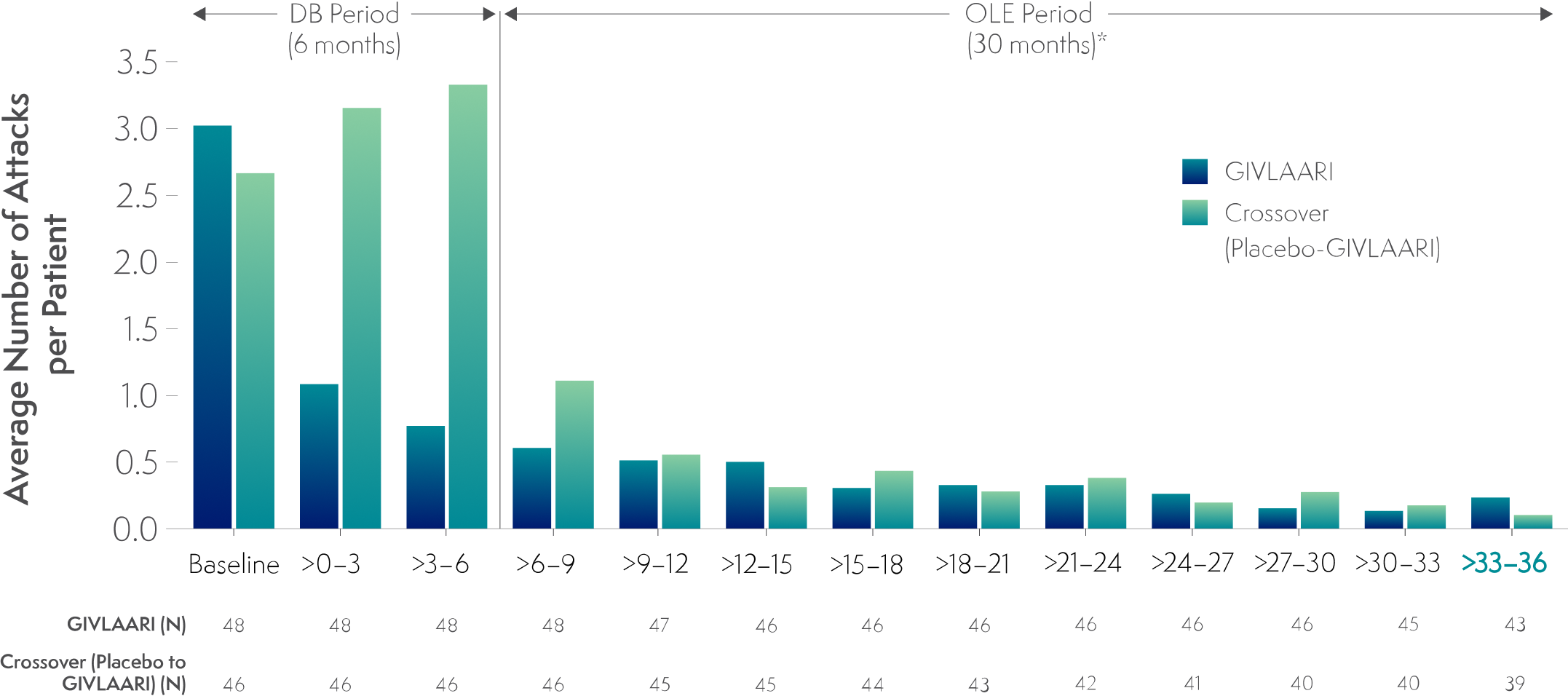
*Data for GIVLAARI 1.25 mg/kg and 2.5 mg/kg in the OLE period were pooled.2
Attacks were defined as those requiring hospitalization, urgent healthcare visit, or intravenous (IV) hemin administration at home.1
- Attack reduction was sustained in patients continuing or crossing over to GIVLAARI treatment during the ENVISION OLE period6
Endpoints in the OLE period are exploratory.6
The average number of attacks per patient in the final 3-month interval of the OLE period was 0.233 and 0.103 in the continuous-GIVLAARI and placebo-crossover groups, respectively5,6
Safety profile of GIVLAARI® (givosiran) in the ENVISION study1
Safety during the 6-month DB period
compared with patients receiving placebo in the 6-month double-blind period1
| Adverse Reaction | GIVLAARI (N=48) n (%) |
Placebo (N=46) n (%) |
| Nausea | 13 (27) | 5 (11) |
| Injection site reactions | 12 (25) | 0 |
| Rash† | 8 (17) | 2 (4) |
| Serum creatinine increase‡ | 7 (15) | 2 (4) |
| Transaminase elevations | 6 (13) | 1 (2) |
| Fatigue | 5 (10) | 2 (4) |
†Grouped term includes pruritus, eczema, erythema, rash, rash pruritic, urticaria.
‡Grouped term includes blood creatinine increased, glomerular filtration rate decreased, chronic kidney disease (decreased estimated glomerular filtration rate).
- Permanent discontinuation of GIVLAARI occurred in 1 patient (2.1%) due to elevated transaminases, in adherence with prespecified protocol-defined stopping rules1
- The most frequently occurring (≥20% incidence) adverse reactions reported in patients treated with GIVLAARI were nausea (27%) and injection site reactions (25%)1
Safety through the OLE period
- The most frequently reported adverse events occurring in ≥20% of patients were injection site reactions, nausea, fatigue, nasopharyngitis, headache, urinary tract infection, and upper respiratory tract infection
- Increased blood homocysteine was reported in 15 of 93 (16%) patients treated with GIVLAARI1
AHP=acute hepatic porphyria; OLE=open-label extension.
In patients with AHP in the ENVISION 6-month double-blind period,
50% were attack-free with GIVLAARI treatment3
Percentage of patients who were attack-free3


Attacks were defined as those requiring hospitalization, urgent healthcare visit, or intravenous (IV) hemin administration at home.1
In a 36-month analysis of patients with AHP,
The number of attack-free patients increased with GIVLAARI treatment6
Percentage of patients who were attack-free* in 3-month† intervals6
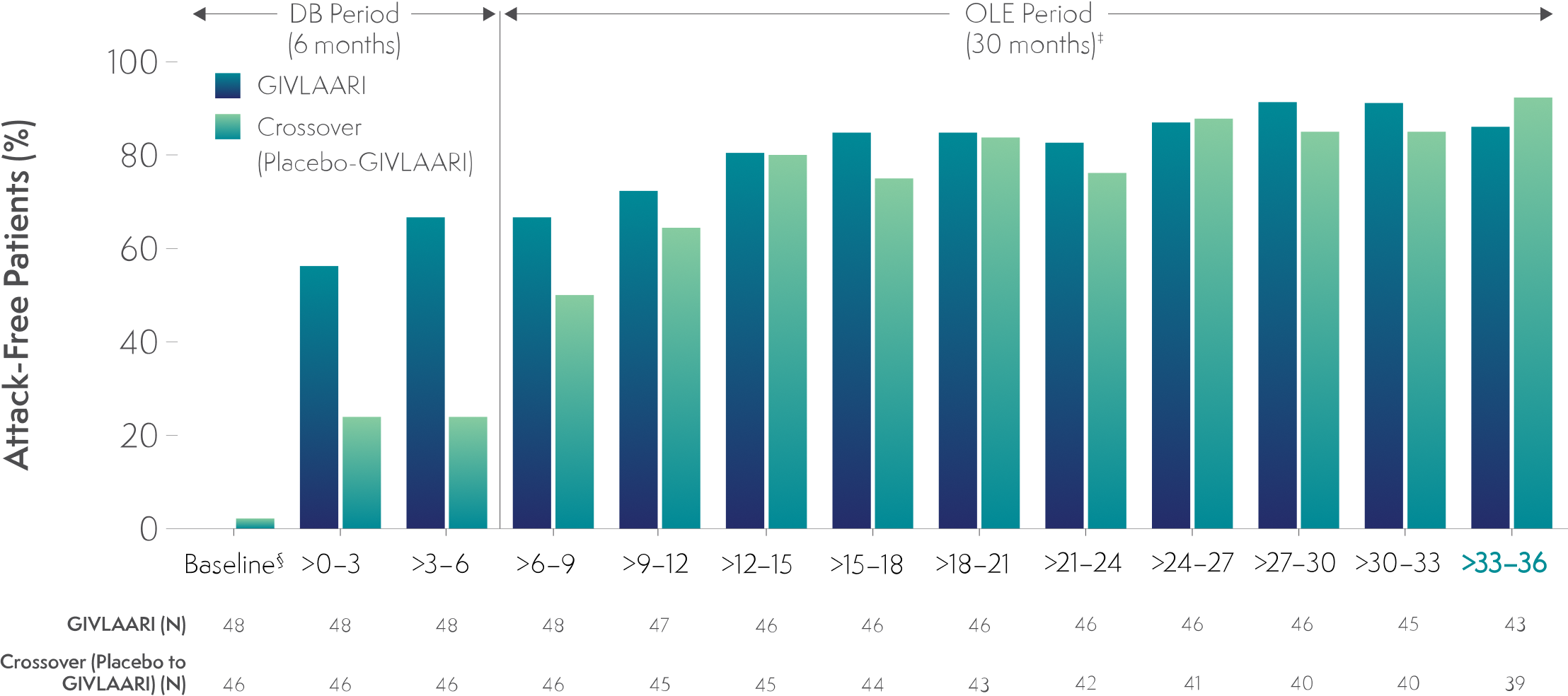
*Attacks were defined as those that required hospitalization, urgent healthcare visit, or intravenous (IV) hemin administration at home.6
†1 month = 28 days.6
‡Data for GIVLAARI 1.25 mg/kg and 2.5 mg/kg in the OLE period were pooled.6
§Baseline represents 6 months before randomization.6
Endpoints in the OLE period are exploratory.6
AHP=acute hepatic porphyria; DB=double-blind; OLE=open-label extension.
In the final 3-month interval of the OLE period, 86% and 92% of patients were attack-free in the GIVLAARI and crossover groups, respectively.‖6
‖Attacks were defined as those requiring hospitalization, urgent healthcare visit, or intravenous (IV) hemin at home.
In patients with AHP in the ENVISION 6-month double-blind period,
GIVLAARI reduced the average days of hemin use by 70%1


- Ratio of average days of hemin use (GIVLAARI:placebo): 0.3 (95% CI: 0.1, 0.5; P=0.0002)1
- In the ENVISION 6-month DB period, 54% of patients with AIP (n=25/46) treated with GIVLAARI had zero days of hemin use compared with 23% of patients (n=10/43) receiving placebo3
AHP=acute hepatic porphyria; AIP=acute intermittent porphyria; DB=double-blind.
In a 36-month analysis of patients with AHP,
Long-term GIVLAARI treatment led to sustained reductions in hemin use6
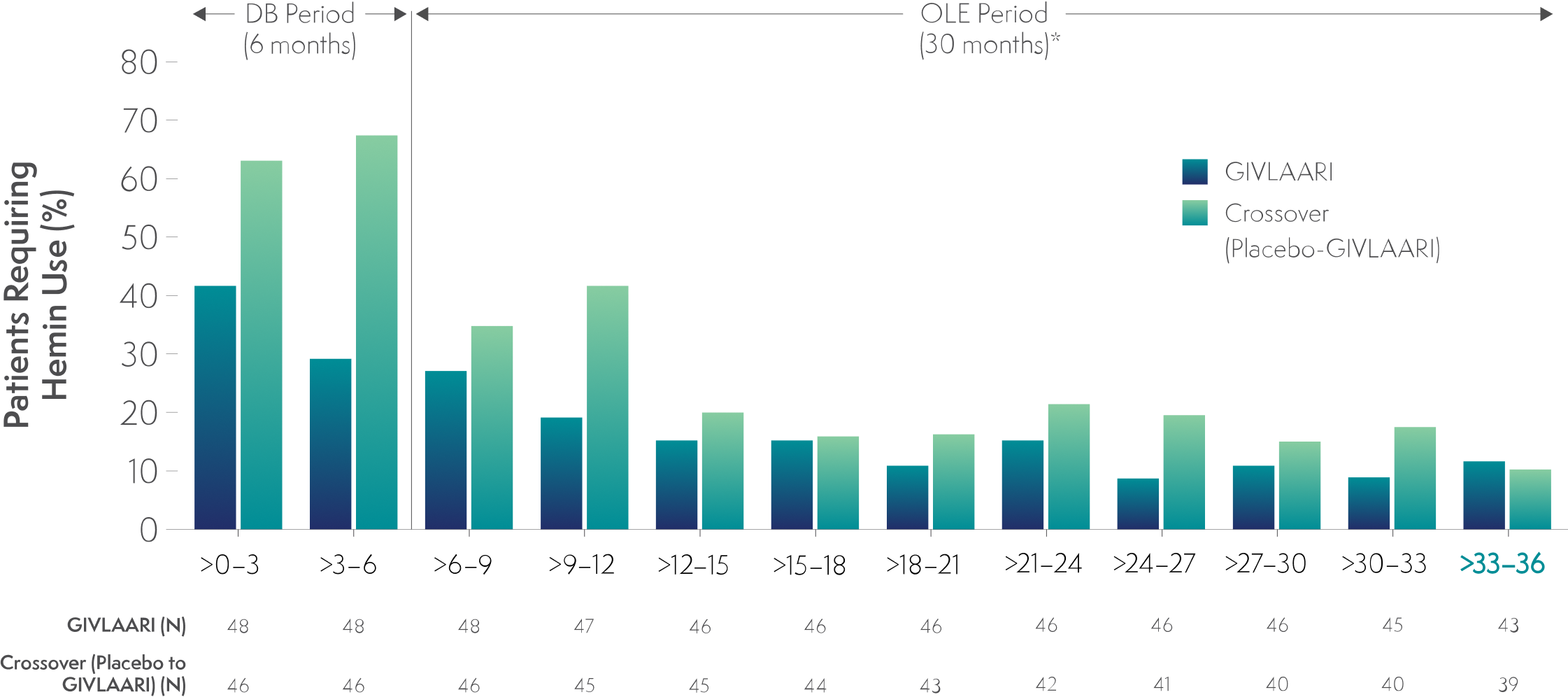
- Patients were required to discontinue prophylactic hemin during the trial6
- 40% of patients with AHP had previous hemin prophylaxis6
- Patients experiencing an AHP attack were treated according to the local standard of care, which could include IV hemin3,6
Endpoints in the OLE period are exploratory.6
In the final 3-month interval of the OLE period, hemin treatment was not required by 88% and 90% of patients in the continuous-GIVLAARI and placebo-crossover groups, respectively6
*Data for GIVLAARI 1.25 mg/kg and 2.5 mg/kg in the OLE period were pooled.6
AHP=acute hepatic porphyria; DB=double blind; OLE=open-label extension.
Daily worst pain scores with GIVLAARI and placebo in patients with AIP3
Daily worst pain score in patients with AIP was a secondary endpoint3
-
Pain was measured using patient-reported daily worst pain scores based on a numeric rating scale (NRS)3
-
A statistical analysis of the change from baseline in daily worst pain score for GIVLAARI vs placebo was conducted3
-
A positive score represents a worsening in daily worst pain from baseline; a negative score represents an improvement from baseline3
-
GIVLAARI did not meet statistical significance for this endpoint based on the prespecified statistical method7
-
The daily worst pain scores were lower with GIVLAARI compared with placebo7
| GIVLAARI (N=46) | Placebo (N=43) | Treatment difference (95% CI) |
| -12.876 | -0.196 | -12.680 (-25.526, 0.166) |
AIP=acute intermittent porphyria; AUC=area under the curve.
Analgesic use with GIVLAARI and placebo in patients with AIP3,8
Analgesic use in patients with AIP was a prespecified exploratory endpoint3,8
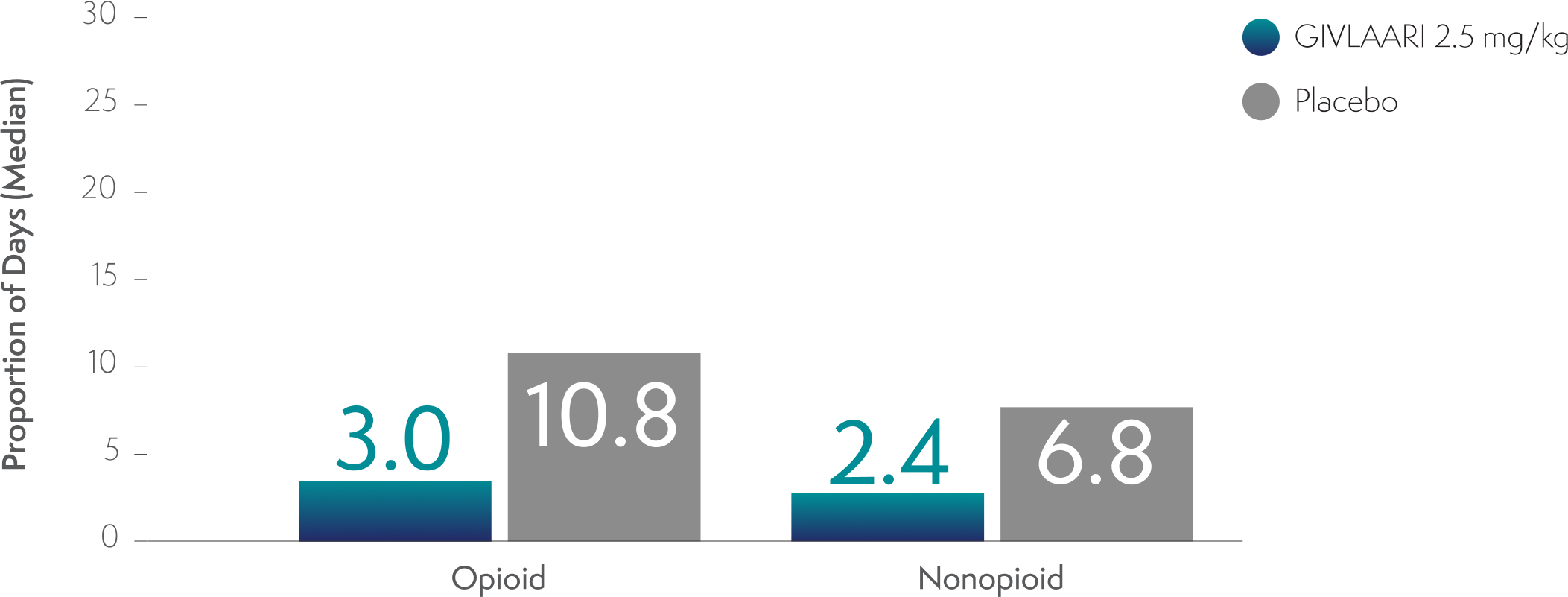
-
Through Month 6 in the double-blind period, the proportion of days with opioid and nonopioid analgesic use was lower in patients treated with GIVLAARI compared with placebo3
-
An evaluation compared with baseline analgesic use cannot be conducted because the proportion of days with analgesic use was not captured at baseline3
AIP=acute intermittent porphyria.
PCS of the SF-12 was evaluated in patients with AIP3
PCS of the SF-12 was viewed as an exploratory endpoint3
- The SF-12 is a 12-question measure capturing global QoL and overall health status3
- Scores on the PCS range from 0 (worst functioning) to 100 (best functioning), with published literature in other chronic diseases suggesting that a change of 2 to 5 points represents a clinically meaningful difference3
- The PCS of the SF-12 was not statistically tested due to not meeting the conditions of the prespecified hierarchical order of statistical testing3,7
- PCS scores were higher in both groups through the OLE period6
- At Month 6 and Month 36, the mean change from baseline in the continuous-GIVLAARI group was 5.1 and 8.6, respectively
- In the placebo-crossover group, the mean change from baseline was 1.7 and 9.4 for these time points, respectively
Limitations
- PCS score included concepts that may not be relevant for the target population (eg, general health, moderate activities, climbing stairs)3
- The domains of bodily pain, social functioning, role limitations due to physical problems, and general health contributed more to the total PCS score3
PCS of SF-12 mean change from baseline to Month 6 and Month 366
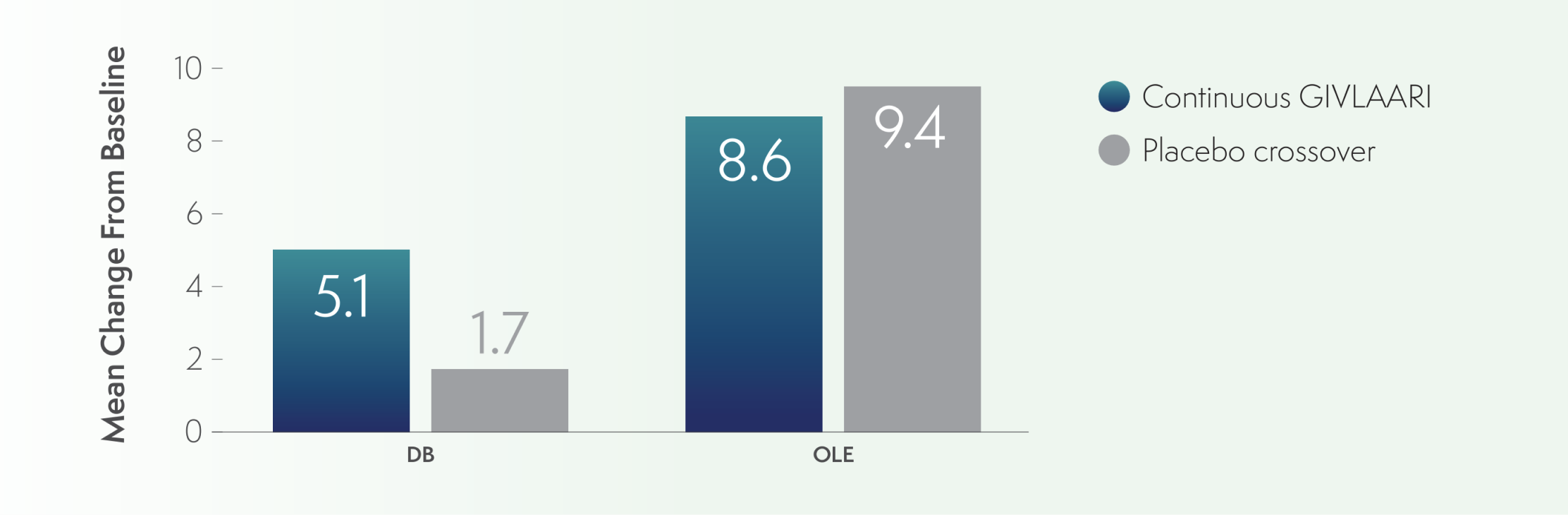
AIP=acute intermittent porphyria; OLE=open-label extension; PCS=Physical Component Summary; QoL=quality of life; SF-12=12-Item Short-Form Health Survey.
Explore the experiences of real patients with AHP taking GIVLAARI
See Their StoriesALAS1=delta-aminolevulinic acid synthase 1; mRNA=messenger RNA.
References: 1. GIVLAARI [prescribing information]. Cambridge, MA: Alnylam Pharmaceuticals, Inc. 2. Ventura P, Bonkovsky HL, Gouya L, et al. Liver Int. 2021;00:1-12. 3. Balwani M, Sardh E, Ventura P, et al; ENVISION Investigators. N Engl J Med. 2020;382:2289-2301. 4. Balwani M, Sardh E, Ventura P, et al. Protocol. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N Engl J Med. 2020;382:2289-2301. 5. Data on file. Alnylam Pharmaceuticals, Inc. 6. Kuter DJ, Bonkovsky HL, Monroy S, et al. J Hepatol. 2023;79(5):1150-1158. 7. Balwani M, Gouya L, Rees DC, et al; ENVISION Investigators. Presented at: EASL International Liver Congress; April 13, 2019; Vienna, Austria. 8. Gouya L, Sardh E, Balwani M, et al. Presented at: ICPP; September 10, 2019; Milan, Italy.